Cell Res:外泌体研究新突破,康铁邦团队发现全新外泌体形成通路
外泌体(Exosome),是细胞外囊泡(EV)的一种主要类型,是直径为30至150 nm的纳米大小的膜结构,大多数细胞都会分泌外泌体。外泌体存在于各种生物体液中,通过其携带的蛋白质、核酸、脂质和代谢物等来发挥细胞间通讯功能,参与免疫应答、病毒感染、代谢和心血管疾病、神经退行性疾病以及癌症进展等多种生理和病理过程。在内体转变为成熟多囊泡内体(MVE)的过程中,内体膜向腔内出芽形成腔内囊泡( ILV),MVE与细胞膜融合释放ILV到细胞外,即形成外泌体。  目前认为内体膜出芽形成ILVs主要通过转运必需内体分选复合体(endosomal sorting complex required for transport, ESCRT),许多熟知的货物蛋白,如蛋白多糖syndecan, 四次跨膜蛋白CD63, 以及 Toll样受体转运伴侣 UNC93B1等,通过细胞质侧的尾巴招募Syntenin-Alix-ESCRT-III通路介导它们的ILVs形成。虽然ESCRT一直被认为是ILV形成所必需的,然而在ESCRT缺失的细胞中,仍然可以观察到MVEs内部存在ILVs,表明细胞中存在ESCRT非依赖的ILV形成方式。 目前认为内体膜出芽形成ILVs主要通过转运必需内体分选复合体(endosomal sorting complex required for transport, ESCRT),许多熟知的货物蛋白,如蛋白多糖syndecan, 四次跨膜蛋白CD63, 以及 Toll样受体转运伴侣 UNC93B1等,通过细胞质侧的尾巴招募Syntenin-Alix-ESCRT-III通路介导它们的ILVs形成。虽然ESCRT一直被认为是ILV形成所必需的,然而在ESCRT缺失的细胞中,仍然可以观察到MVEs内部存在ILVs,表明细胞中存在ESCRT非依赖的ILV形成方式。
2020年9月22日,中山大学肿瘤防治中心康铁邦团队在Cell Research 在线发表了文题为:RAB31 marks and controls an ESCRT-independent exosome pathway 的研究论文。 在这项研究中,康铁邦团队鉴定到一条由RAB31标记并控制的ESCRT非依赖的外泌体通路,极大地提升了我们对外泌体生物发生的认识和理解。

 外泌体的生物发生途径主要包括三个关键的检查点:ILV的形成,阻止MVEs的降解以及MVEs和细胞膜的融合,这三个检查点都包含在内体相关的囊泡运输过程中。RAB GTPase定位到特定膜结构的表面,通过招募效应因子来调节相应膜结构的囊泡运输,例如,在内体溶酶体运输网络中,RAB5调节早期内体的形成及相互融合;内体膜上RAB5到RAB7的转换调节早期向晚期内体的转变;RAB7调节晚期内体/MVEs与溶酶体的融合来降解ILVs;RAB27调节MVEs与细胞膜的对接和融合来释放ILVs形成外泌体。内吞的膜蛋白,特别是受体酪氨酸激酶(receptor tyrosine kinase, RTK)家族的表皮生长因子受体(epidermal growth factor receptor, EGFR),定位到内体和MVEs,通过MVEs和溶酶体融合来进入溶酶体降解,此过程受多种RAB GTPases和ESCRT复合体的调控。 外泌体的生物发生途径主要包括三个关键的检查点:ILV的形成,阻止MVEs的降解以及MVEs和细胞膜的融合,这三个检查点都包含在内体相关的囊泡运输过程中。RAB GTPase定位到特定膜结构的表面,通过招募效应因子来调节相应膜结构的囊泡运输,例如,在内体溶酶体运输网络中,RAB5调节早期内体的形成及相互融合;内体膜上RAB5到RAB7的转换调节早期向晚期内体的转变;RAB7调节晚期内体/MVEs与溶酶体的融合来降解ILVs;RAB27调节MVEs与细胞膜的对接和融合来释放ILVs形成外泌体。内吞的膜蛋白,特别是受体酪氨酸激酶(receptor tyrosine kinase, RTK)家族的表皮生长因子受体(epidermal growth factor receptor, EGFR),定位到内体和MVEs,通过MVEs和溶酶体融合来进入溶酶体降解,此过程受多种RAB GTPases和ESCRT复合体的调控。
ESCRT分选泛素化的EGFR进入MVEs形成ILVs来促进EGFR的溶酶体降解,是MVE通路上膜蛋白分选进入内体溶酶体降解的经典模型。事实上,EGFR在多种癌症中经常积累并伴随着突变,并且存在于癌细胞系和癌症病人血清来源的外泌体中。这个现象表明分选EGFR进入ILVs走向外泌体分泌的通路可能不同于ESCRT复合体,体现在ILV形成和阻止MVE降解这两个关键检查点,极有可能受RAB GTPase的调控。
为了找到控制EGFR外泌体形成的RAB GTPase,研究团队首先构建了一个由62个RAB GTPases组成的激活型RAB GTPase文库,在正常培养条件下的HeLa细胞中,利用免疫荧光筛选到激活型RAB31 (RAB31Q65L)指导EGFR定位到膨大的CD63阳性MVEs (图1)。研究团队进一步发现在血清饥饿条件下,非泛素化的EGFR依然会内吞进入细胞,而RAB31Q65L驱动内吞的EGFR定位到膨大的CD63阳性MVEs而不进入溶酶体。结构化照明显微镜(SIM)和3D-SIM的结果显示RAB31Q65L和EGFR进入CD63阳性MVEs内部并定位到ILVs结构,免疫电镜的结果清晰地显示它们定位到ILV的膜结构上(图1)。
进一步地,研究团队发现RAB31Q65L显著增加浓缩条件培养基(concentrated conditional media, CCM)中的EGFR(图1),以及一些熟知的EV标记物,如FLOT1、 FLOT2、CD9、CD81和CD63,但是ESCRT相关的EV标记物,如Tsg101和Alix,并没有改变,暗示ESCRT可能不参与RAB31Q65L驱动的EGFR外泌体的形成。确实,研究团队进一步通过敲降ESCRT组分Hrs和Tsg101,以及相关蛋白Alix,证明它们不参与RAB31Q65L驱动的EGFR外泌体的形成。因此,研究团队提出激活型RAB31标记并控制一条ESCRT非依赖的外泌体通路。
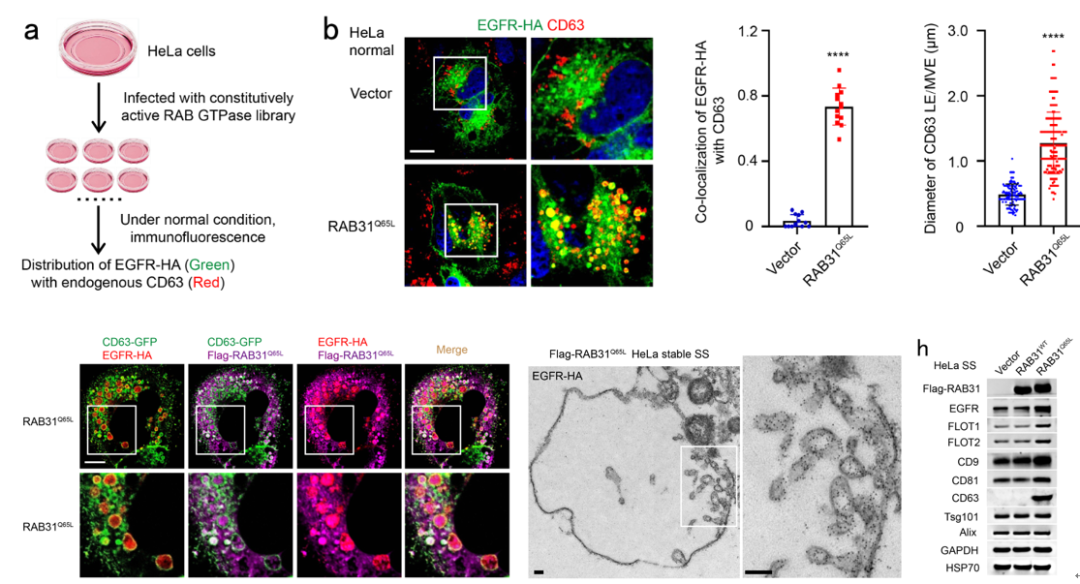 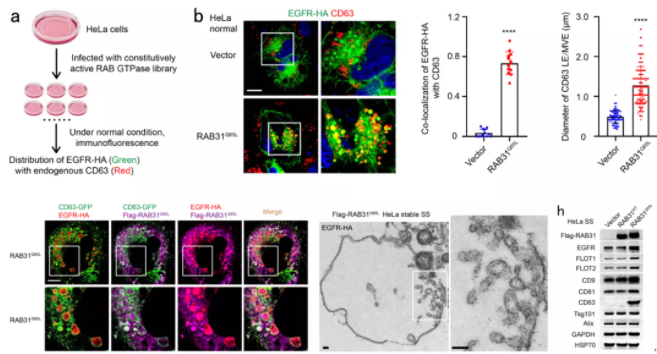
图1 RAB31Q65L驱动EGFR进入MVEs形成ILVs和外泌体
进一步,研究团队对RAB31Q65L驱动EGFR外泌体形成的效应因子和机制进行探究。值得注意的是,作为内体膜和细胞膜的脂筏结构蛋白FLOT1和FLOT2,同时也作为经典的EV标记物,在RAB31Q65L来源的CCM中高度富集。通过一系列基因敲降、抑制剂处理、免疫荧光和外泌体组分分析,研究团队发现FLOTs是RAB31Q65L驱动EGFR外泌体形成的效应因子,脂筏结构的组分胆固醇和神经酰胺是RAB31Q65L-FLOTs诱导MVE膜出芽形成ILVs所必需的。通过截短回补实验,研究团队发现激活型RAB31结合FLOTs的SPFH结构域并通过Flotillin结构域来诱导MVEs膜出芽形成ILVs。
进一步,研究团队对激活RAB31的上游调控信号进行探寻。血清饥饿条件下,RAB31WT只能指导EGFR定位到晚期内体的膜上,而不进入MVEs内部。有趣的是,EGF刺激可以促进RAB31WT和EGFR进入CD63阳性MVEs内部,同时促进EGFR外泌体的产生。接着,研究团队发现EGFR通过磷酸化RAB31的Y76、Y86和Y137三个位点的酪氨酸,使其具有和激活型RAB31Q65L相同的功能。这三个酪氨酸位点的突变体RAB313YF不能驱动EGFR进入MVEs内部,同时显著减少EGFR外泌体的产生。研究团队进一步发现多种RTKs可以通过磷酸化RAB31使其转变为激活型,接着激活型RAB31通过脂筏微结构域中的FLOTs驱动这些RTKs进入MVEs形成ILVs及相应的外泌体。进一步,研究团队通过对CCM中外泌体蛋白组分的检测和分析来比较两条外泌体通路的关系,发现RAB31-FLOTs和Syntenin-Alix-ESCRT-III通路是两条平行的外泌体形成通路,分别负责不同的货物。
最后,研究团队对RAB31能否阻止MVEs的降解进行探究。在EGF刺激条件下,通过免疫荧光和免疫印迹追踪EGFR的定位和降解,发现外源表达的RAB31可以将EGFR阻滞在CD63阳性MVEs而不进入溶酶体,即RAB31通过抑制MVEs和溶酶体融合来阻止EGFR的溶酶体降解。因为MVEs和溶酶体融合由激活型RAB7介导,研究团队推测高表达的RAB31会抑制RAB7的活性。通过检测细胞中激活型RAB7的水平和定位,研究团队证实外源表达的RAB31会显著降低细胞中激活型RAB7的水平,而且RAB31定位的晚期内体和MVEs表面没有激活型RAB7的分布。进一步,研究团队发现RAB31招募GTP酶激活蛋白(GTPase-activating protein, GAP) TBC1D2B到晚期内体/MVEs表面来失活RAB7。此外,内源RAB31的敲降不仅会导致TBC1D2B不再定位到晚期内体/MVEs,而且会显著地提升细胞中激活型RAB7的水平。
总的来说,该研究揭示了RAB31在外泌体生物发生过程中的双重功能:驱动ILVs的形成和阻止MVEs的降解。多种RTKs通过磷酸化来激活RAB31,激活型RAB31通过结合脂筏微结构域中的FLOTs蛋白驱动MVEs膜出芽形成ILVs,同时RAB31招募TBC1D2B到MVEs表面失活RAB7来抑制MVEs和溶酶体的融合,从而有利于MVEs和细胞膜融合释放ILVs形成外泌体(图2)。
此外,该研究对外泌体生物发生相关分子机器的重新梳理和定义,为更好地理解外泌体生物发生的复杂异质性迈出了关键一步。对RAB31相关分子机器的深入理解,可为多种人类疾病(如癌症和神经退行性疾病等)治疗策略的设计提供重要的理论基础。
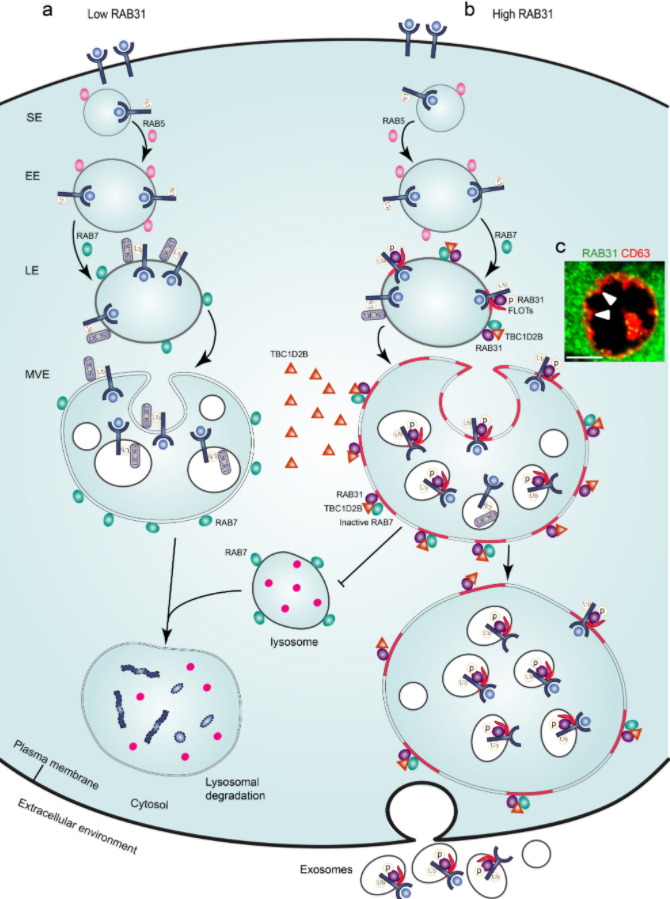 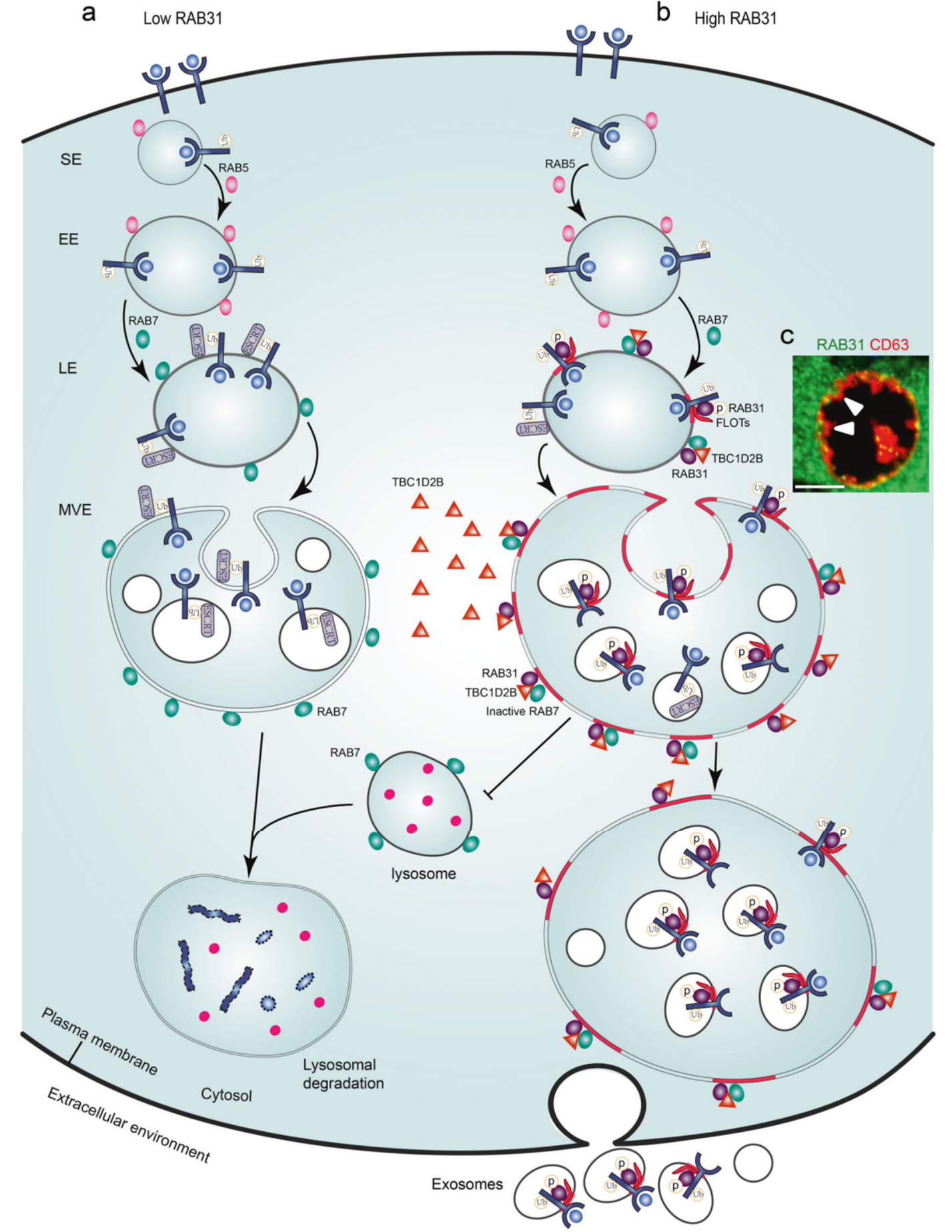
图2 RAB31标记并控制外泌体通路的作用模式图
据悉,中山大学肿瘤防治中心魏灯辉助理研究员、詹伟祥博士和高瑛博士为共同第一作者;中山大学肿瘤防治中心康铁邦教授为通讯作者。
附件已隐藏,回复该贴可查看附件
| 


 /1
/1 
 |Archiver|手机版|外泌体之家 | exosomes & microvesicles
|Archiver|手机版|外泌体之家 | exosomes & microvesicles







 发表于 2020-10-16 20:26:58
发表于 2020-10-16 20:26:58

 收藏
收藏